1. Sequence Alignment/Map Format Specification
The official defination is here.
“It is a TAB-delimited text format consisting of a header section, which is optional, and an alignment section. If present, the header must be prior to the alignments. Header lines start with ‘@’, while alignment lines do not. Each alignment line has 11 mandatory fields for essential alignment information such as mapping position, and variable number of optional fields for flexible or aligner specific information.”
The SAM, VCF, GFF and Wiggle formats are using the 1-based coordinate system.
The BAM, BCFv2, BED, and PSL formats are using the 0-based coordinate system.
1.1 The header section
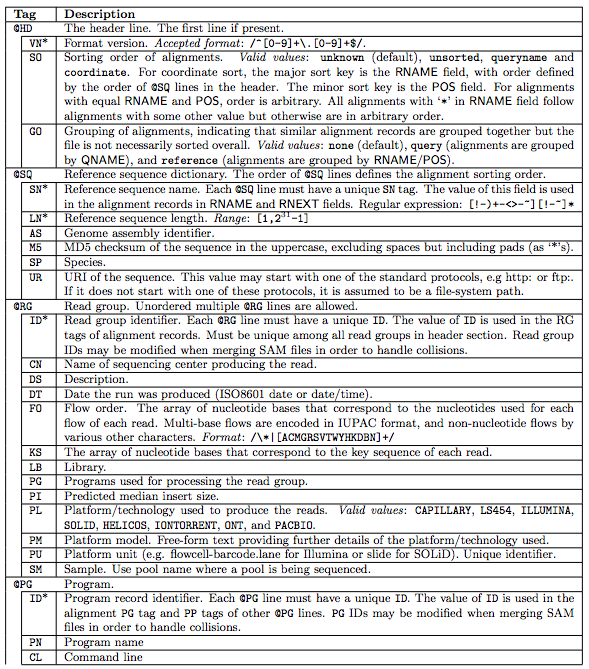

1.2 The alignment section: mandatory fields
“In the SAM format, each alignment line typically represents the linear alignment of a segment. Each line has 11 mandatory fields. These fields always appear in the same order and must be present, but their values can be ‘0’ or ‘*’ (depending on the field) if the corresponding information is unavailable. The following table gives an overview of the mandatory fields in the SAM format”

1.2.1 The FLAG field

1.2.2 The CIGAR field

2. BAM format
SAM files and BAM files contain the same information, but in a different format. BAM is compressed in the BGZF format.
3. Practice
3.1 How many alignments does the BAM file contain?
A BAM file contains alignments for a set of input reads. Each read can have 0 (none), 1 or multiple alignments on the genome. The number of alignments is the number of entries, excluding the header, contained in the BAM file, or equivalently in its SAM conversion.
samtools flagstat test.bam
An alternate method would be to count the number of lines in the converted SAM file (header excluded):
samtools view test.bam | wc -l
If the BAM file was created with a tool that includes unmapped reads into the BAM file, we would need to exclude the lines representing unmapped reads, i.e. with a “*” in column 3 (chrom)
samtools view test.bam | cut -f 3 | grep -v '*' | wc -l
3.2 How many alignments show the read’s mate unmapped?
An alignment with an unmapped mate is marked with a ‘*’ in column 7.
samtools view test.bam | cut -f 7 | grep -c '*'
3.3 How many alignments contain a deletion (D)?
Deletions are be marked with the letter ‘D’ in the CIGAR string for the alignment, shown in column 6.
samtools view test.bam | cut -f 6 | grep -c 'D'
3.4 How many alignments show the read’s mate mapped to the same chromosome?
An alignment with mate mapped to same chromosome is marked with a “=” in column 7.
samtools view test.bam | cut -f 7 | grep -c '='
3.5 How many alignments are spliced?
A spliced alignment will be marked with an “N” (intron gap) in the CIGAR field (column 6).
samtools view test.bam | cut -f 6 | grep -c 'N'
3.6 How many sequences are in the genome file?
This information can be found in the header of the BAM file. The number of lines describing the sequences in the reference genome.
samtools view -H test.bam | grep -c "SN:"
3.7 What is the length of the first sequence in the genome file?
The length information is stored alongside the sequence identifier in the header (pattern “LN:seq_length”).
samtools view -H test.bam | grep "SN:" | more
3.8 What alignment tool was used?
The program name is listed in the @PG line in the BAM header (pattern “ID:program_name”).
samtools view -H test.bam | grep "^@PG"
3.9 Extract a subregion from the BAM file.
Extract 1,000,000 to 10,000,000 on chromsome 3.
echo "Chr3 1000000 10000000" > region.bed
samtools view -b -L region.bed test.bam > test_region.bam